|
Modelagem e Simulação de Processos Optoeletrônicos em Semicondutores Orgânicos |
|
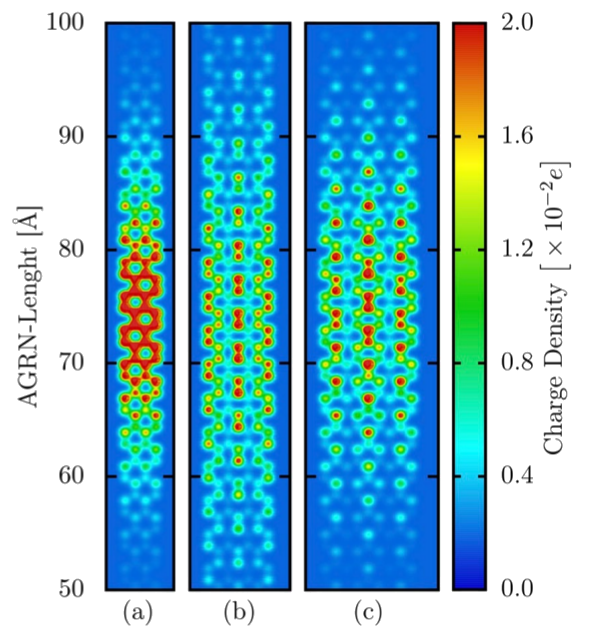
| Por meio de métodos de química quântica e física computacional, este projeto se propõe à modelagem computacional de diversos sistemas orgânicos a serem utilizados como camadas ativas em dispositivos optoeletrônicos, especialmente em sistemas fotovoltaicos. A investigação de processos de transferência, separação e recombinação de portadores de carga em heterojunções orgânicas, sistemas que podem ser usados no desenvolvimento de células solares (dispositivos fotovoltaicos), será o principal objeto de estudo. Considerável parte deste projeto é dedicada ao desenvolvimento de novas metodologias capazes de tornar as condições de simulação mais realistas e os resultados mais acurados, abrangendo, portanto, um viés metodológico. O desenvolvimento dessas novas metodologias pode proporcionar uma descrição mais detalhada e precisa da estrutura eletrônica de vários sistemas orgânicos, tanto em nível atômico quanto na escala molecular, contribuindo para um melhor entendimento dos processos físicos envolvidos no funcionamento dos dispositivos optoeletrônicos e, também, para o desenvolvimento de novos materiais, o que é muito atrativo dos pontos de vista acadêmico e industrial. Cálculos de dinâmica molecular serão empregados para estudar heterojunções orgânicas em candidatos a sistemas fotovoltaicos de alto desempenho. Combinando os cálculos de dinâmica molecular e mecânica quântica, será possível propor uma descrição mais realista dos problemas de transporte, transferência, separação e recombinação de carga em várias classes de condutores orgânicos. Também, entre os problemas estudados, destaca-se a dinâmica de estados excitados, como o transporte de polarons, por exemplo, em polímeros conjugados e em nanofitas de grafeno. O problema da dinâmica de estados excitados em sistemas orgânicos é investigado no escopo de modelos Tight-Binding (em português Ligação Rígida) com relaxação em uma e duas dimensões. | |
|
Publicações Relacionadas: Advanced Theory and Simulations, 2200877, 2023. Journal of Materials Chemistry C, 7, 4066-4071, 2019. Journal of Physical Chemistry Letters, 6, 510-514, 2015. Journal of Physical Chemistry Letters, 20, 3039-3042, 2012.
Pessoas para Contato: Marcelo Lopes Pereira Junior Luiz Antonio Ribeiro Junior Pedro Henrique de Oliveira Neto |
 |
______________________________________________________________________________________________________________________
| Estrutura e Transporte Eletrônico em Redes de Baixa Dimensionalidade | |
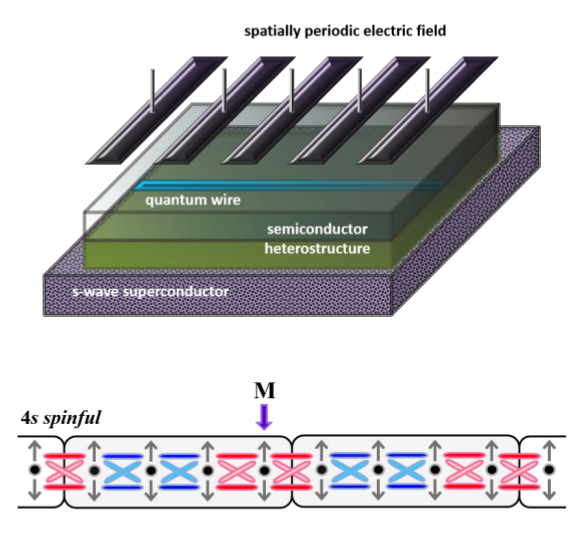
| Sistemas quânticos fortemente correlacionados em baixas dimensões que apresentam novos tipos de transições de fase quânticas e fases topológicas. Temos investigado cadeias de spin, lı́quidos de Luttinger, fios quânticos com interação spin-órbita na presença ou não de supercondutividade, sistemas eletrônicos com simetrias especiais, e modelos quânticos com anomalias termodinâmicas. A abordagem teórica destes sistemas envolve teoria de bandas topológica, grupo de renormalização, bosonização, técnicas perturbativas e diagramáticas em teoria de muitos corpos, teoria quântica de campos e métodos computacionais. | |
|
Publicações Relacionadas: Phys. Rev. B 105, 155414, 2022. Physical Review B, 98, 165127, 2018. Physical Review B, 94, 115128, 2016. Physica B-Condensed Matter, 494, 1-6, 2016.
Pessoas para Contato: Mariana Malard Paulo Eduardo de Brito
|
 |
______________________________________________________________________________________________________________________
| Propriedades Anômalas da Água | |
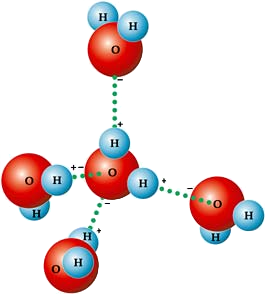
| Além de ser um líquido relevante devido ao ser caracter único para a e existência e mautenção da vida, a água também é importante em processos industriais, no transporte e em diversos setores econômicos em nossa sociedade. Do ponto de vista científico a água tem sido objeto de estudo intenso em diversas áreas, considerando que que possui mais de 70 comportamentos considerados anômalos. Neste projeto investigamos comportamentos termodinâmicos e cinéticos da água buscando, por exemplo, entender a relação entre a anomalia na densidade, presente entre 0 e 4ºC a pressão ambiente, funções termodinâmicas de resposta e transições de fase líquido-líquido. Para isso utilizamos métodos teóricos (Rede de Bethe, Técnica da Matriz de Transferência, várias metodologias de campo médio) e computacionais (Simulações de Monte e Dinâmica Molecular). Entre nossos principais resultados propusemos uma relação entre o fenômeno de entropia residual e o aparecimento de anomalias típicas da água em sistemas em rede, o que permitiu, recentemente, prever que gases quânticos ultra-resfriados em redes óticas apresentam propriedades anômalas similares à água. | |
|
Publicações Relacionadas: Phys. Rev. E 105, 044110, 2022. Frontiers of Physics, 3, 136102, 2017. Physical Review E, 87, 032303, 2013.
Pessoas para Contato: Marco Aurélio Barbosa Fernandes Mariana Malard
|
 |
______________________________________________________________________________________________________________________
|
Uso da Dinâmica Molecular Reativa para o Estudo das Propriedades Físico-Químicas de Novas Nanoestruturas |
|
|
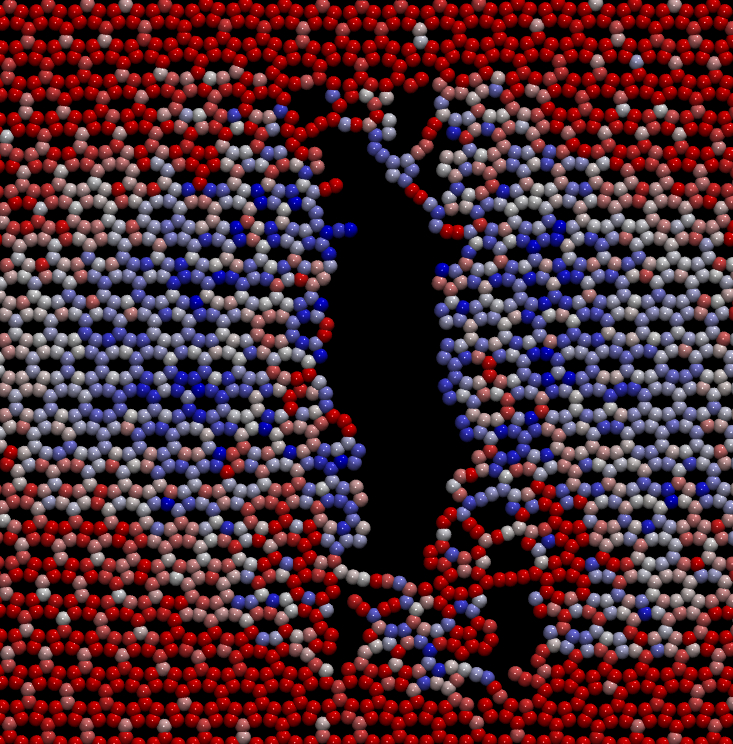
Esta linha se destina ao estudo das propriedades físico-químicas de nanoestruturas baseadas em novos alótropos de carbono, buscando propor materiais mais eficientes no que tange às aplicações de conversão e armazenamento de energia. Entre os problemas estudados destacam-se as propriedades estruturais, mecânicas, padrões de fraturas e degradação em atmosferas gasosas destes sistemas nas formas de monocamada, tubo e scroll. Considerável parte desse projeto é dedicada ao desenvolvimento de novas metodologias, baseadas em dinâmica molecular reativa, capazes de tornar as condições de simulação mais realistas e os resultados mais acurados para uma boa descrição dos dados fornecidos pelos experimentos na literatura. A dinâmica molecular, com o uso de um potencial reativo, permite estudar a dissociação e a formação de ligações químicas em sistemas nanoestruturados. O desenvolvimento dessas novas metodologias baseadas em dinâmica molecular pode proporcionar elementos para uma descrição mais detalhada e precisa da estrutura eletrônica desses sistemas tanto em nível atômico quanto na escala molecular, contribuindo para um melhor entendimento dos processos físico-químicos envolvidos no funcionamento de dispositivos optoeletrônicos, também, para o desenvolvimento de novos materiais, o que é muito atrativo dos pontos de vista acadêmico e industrial. Dentre as ferramentas computacionais utilizadas nessa linha de pesquisa destacam-se: LAMMPS (potenciais ReaxFF, AIREBO, Tersoff e Stillinger-Weber (SW)) e Materials Studio (Módulo GULP). |
|
|
Publicações Relacionadas: Carbon 189 (2022): 422-429. J. Phys. Chem. C 2020, 124, 27, 14855-14860 Chemical Physics Letters, 756, 2020, 137830 Marcelo Lopes Pereira Junior Luiz Antonio Ribeiro Junior
|
 |